Cellular senescence is the phenomenon where cells lose the ability to divide. In response to DNA damage (including shortened telomeres) cells either senesce or self-destruct (apoptosis) if the damage cannot be repaired. Organismal senescence is the aging of whole organisms. The term aging has become so commonly equated with senescence that the terms will be used interchangeably in this article. Senescence is studied in gerontology which is the branch of science involved with the aging process.
The process of senescence is complex, and may derive from a variety of different mechanisms and exist for a variety of different reasons. However, senescence is not universal, and scientific evidence suggests that cellular senescence evolved in certain species as a mechanism to prevent the onset of cancer. Moreover senescence is negligible and cannot be detected in many organisms, including sponges, corals, and lobsters. All such species have no “post-mitotic” cells; they reduce the effect of damaging free radicals by cell division and dilution. Such species are not immortal, however, as they will eventually fall prey to trauma or disease. Moreover, average lifespans can vary greatly within and between species. This suggests that both genetic and environmental factors contribute to aging.
How and why some cells become post-mitotic in some species has been the subject of much research and speculation, but (as noted above) it is widely believed that cellular senescence evolved as a way to prevent the onset and spread of cancer. Somatic cells that have divided many times will have accumulated DNA mutations and would therefore be in danger of becoming cancerous if cell division continued.

Lately the role of telomeres in cellular senescence has aroused general interest, especially with a view to the possible genetically adverse effects of cloning. The successive shortening of the chromosomal telomeres with each cell cycle is also believed to limit the number of divisions of the cell, thus contributing to aging. There have, on the other hand, also been reports that cloning could alter the shortening of telomeres. Some cells do not age and are therefore described as being “biologically immortal.” It is theorized by some that when it is discovered exactly what allows these cells, whether it be the result of telomere shortening or not, to divide without limit that it will be possible to genetically alter other cells to have the same capability. It is further theorized that it will eventually be possible to genetically engineer all cells in the human body to have this capability by employing gene therapy and thereby stop or reverse ageing, effectively making the entire organism potentially immortal.
Theories that explain senescence can generally be divided between the programmed and error theories of aging.
Programmed theories imply that aging is regulated by biological clocks operating throughout the life span. This regulation would depend on changes in gene expression that affect the systems responsible for maintenance, repair and defense responses.
Error theories blame environmental impacts on living organisms that induce cumulative damage at various levels as the cause of aging, examples which range from damage to deoxyribonucleic acid (DNA), damage to tissues and cells by oxygen radicals (widely known as free radicals countered by the even more well known antioxidants), and cross-linking.
Evolutionary theories
Evolutionary theory explains why we don’t live forever, and provides a reasonable explanation for the huge variation in lifespan between (often closely related) species.
The geneticist J. B. S. Haldane wondered why the dominant mutation which causes Huntington’s disease remained in the population, why natural selection had not eliminated it. The onset of this neurological disease is (on average) at age 35 and is invariably fatal within 10-20 years. Haldane assumed, probably reasonably, that in human prehistory, few survived until age 35. Since few were alive at older ages and their contribution to the next generation was therefore small relative to the large cohorts of younger age groups, the force of selection against such late-acting deleterious mutations was correspondingly small. If a mutation affected younger individuals, selection against it would be strong. Hence, late-acting deleterious mutations could accumulate in populations over evolutionary time through genetic drift. This principle has been proven correct.
Peter Medawar formalized this observation in his Mutation Accumulation theory of aging. “The force of natural selection weakens with increasing age – even in a theoretically immortal population, provided only that it is exposed to real hazards of mortality. If a genetic disaster… happens late enough in individual life, its consequences may be completely unimportant”. The ‘real hazards of mortality’ are typically predation, disease and accidents. So, even an immortal population, whose fertility does not decline with time, will have fewer individuals alive in older age groups. This is called ‘extrinsic mortality’. Young age cohorts, not depleted in numbers yet by extrinsic mortality, contribute far more to the next generation than the few remaining in older age cohorts, so the force of selection against late-acting deleterious mutations, which only affect these few older individuals, is very weak.
The major testable prediction made by this model is that species which have high extrinsic mortality in nature will age more quickly and have shorter lifespans, and it is borne out among mammals, the most well studied in terms of life history. There is a correlation among mammals between body size and lifespan, such that larger species live longer than smaller species in controlled/optimum conditions, but there are notable exceptions. For instance, many bats and rodents are similarly sized, yet bats live much, much longer. For instance, the little brown bat, half the size of a mouse, can live 30 years in the wild. A mouse will live 2-3 years even with optimum conditions. The explanation is that bats have fewer predators, therefore low extrinsic mortality. Thus more individuals survive to later ages so the force of selection against late-acting deleterious mutations is stronger. Fewer late-acting deleterious mutations = slower aging = longer lifespan. Birds are also warm-blooded and similarly sized to many small mammals, yet live often 5-10 times as long. They clearly have fewer predation pressures compared with ground-dwelling mammals. And sea-birds, which generally have the fewest predators of all birds, live longest.
Also, when examining the body-size vs. lifespan relationship, predator mammals tend to have longer lifespans than prey animals in a controlled environment such as a zoo or nature reserve. The explanation for the long lifespans of primates (such as humans and monkeys and apes) relative to body size is that their intelligence and often sociality helps them avoid becoming prey.
Another evolutionary theory of aging was proposed by George C. Williams (Williams 1957) and involves antagonistic pleiotropy. A single gene may effect multiple traits. Some traits that increase fecundity early in life may also have negative effects later in life. Williams suggested the following example: perhaps a gene codes for calcium deposition in bones which promotes juvenile survival and will therefore be favored by natural selection; however this same gene promotes calcium deposition in the arteries, causing negative effects in old age. Therefore negative effects in old age may reflect the result of natural selection on pleiotropic genes early in life.
Ultimately, lifespan differences among species are due to genetics, but this does not explain the “how” question of aging.
Gene regulation
A number of genetic components of aging have been identified using model organisms, ranging from the simple budding yeast Saccharomyces cerevisiae to worms such as Caenorhabditis elegans and fruit flies. Study of these organisms has revealed the presence of at least two conserved aging pathways.
One of these pathways involves the gene Sir2, a NAD+-dependent histone deacetylase. In yeast, Sir2 is required for genomic silencing at three loci: the yeast mating loci, the telomeres and the ribosomal DNA (rDNA). Yeast replicative aging is caused by homologous recombination between rDNA repeats; excision of rDNA repeats results in the formation of extrachromosomal rDNA circles (ERCs). These ERCs replicate and preferentially segregate to the mother cell during cell division, and are believed to result in cellular senescence by titrating away (competing for) essential nuclear factors. While ERCs are not believed to contribute to aging in higher organisms such as human, extrachromosomal circular DNA (eccDNA) has been found in worms, flies and humans. The role of eccDNA in aging is unknown.
Despite the lack of a connection between circular DNA and aging in higher organisms, extra copies of Sir2 are capable of extending the lifespan of both worms and flies. The mechanisms by which Sir2 homologues in higher organisms regulate lifespan is unclear, but the human SIRT1 protein has been demonstrated to deacetylate p53, Ku70, and the forkhead family of transcription factors. SIRT1 can also regulate acetylates such as CBP/p300, and has been shown to deacetylate specific histone residues.
Other genes regulate aging in yeast by increasing the resistance to oxidative stress. Superoxide dismutase, a protein that protects against the effects of mitochondrial free radicals, can extend yeast lifespan in stationary phase when overexpressed.
In higher organisms, aging is likely to be regulated in part through the insulin/IGF-1 pathway. Mutations that affect insulin-like signaling in worms, flies and mice are associated with extended lifespan. In yeast, Sir2 activity is regulated by the nicotinamidase PNC1. PNC1 is transcriptionally upregulated under stressful conditions such as caloric restriction, heat shock, and osmotic shock. By converting nicotinamide to nicotinic acid, it removes nicotinamide, which inhibits the activity of Sir2. A nicotinamidase found in humans, known as PBEF, may serve a similar function, and a secreted form of PBEF known as visfatin may help to regulate serum insulin levels.
It is not known, however, whether these mechanisms also exist in humans since there are obvious differences in biology between humans and model organisms.
Chemical damage
It is also suggested that damage to long-lived biopolymers, such as structural proteins or DNA, caused by ubiquitous chemical agents in the body such as oxygen and sugars, are in part responsible for aging. The damage can include breakage of biopolymer chains, cross-linking of biopolymers, or chemical attachment of unnatural substituents (haptens) to biopolymers.
Under normal aerobic conditions, approximately 4% of the oxygen metabolized by mitochondria is converted to superoxide ion which can subsequently be converted to hydrogen peroxide, hydroxyl radical and eventually other reactive species including other peroxides and singlet oxygen, which can in turn generate free radicals capable of damaging structural proteins and DNA. Certain metal ions found in the body, such as copper and iron, may participate in the process. (In Wilson’s disease, a hereditary defect which causes the body to retain copper, some of the symptoms resemble accelerated senescence.) These processes are termed oxidant damage and are the target of the currently popular nutritional antioxidants.
Sugars such as glucose and fructose can react with certain amino acids such as lysine and arginine and certain DNA bases such as guanine to produce sugar adducts, in a process called glycation. These adducts can further rearrange to form reactive species which can then cross-link the structural proteins or DNA to similar biopolymers or other biomolecules such as non-structural proteins. People with diabetes, who have elevated blood sugar, develop senescence-associated disorders much earlier than the general population, but can delay such disorders by rigorous control of their blood sugar levels. There is evidence that sugar damage is linked to oxidant damage in a process termed glycoxidation.
Free radicals can damage proteins, lipids or DNA. Glycation mainly damages proteins. Damaged proteins and lipids accumulate in lysosomes as lipofuscin. Chemical damage to structural proteins can lead to loss of function; for example, damage to collagen of blood vessel walls can lead to vessel-wall stiffness and thus hypertension, and vessel wall thickening and reactive tissue formation (atherosclerosis); similar processes in the kidney can lead to renal failure. Damage to enzymes reduces cellular functionality. Lipid peroxidation of the inner mitochondrial membrane reduces the electric potential and the ability to generate energy. It is probably no accident that almost all of the so-called “accelerated aging diseases” are due to defective DNA repair enzymes.
Neuro-endocrine-immunological theories
Senescence may also simply be a result of wear and tear overwhelming repair mechanisms. It is also possible that senescence is a mechanism to control the development and spread of cancer; if cells have built-in limits to how many times they can replicate, they must somehow overcome this before they can spread indefinitely.
Miscellaneous
Recently, early senescence has been alleged to be a possible unintended outcome of early cloning experiments, Most notably, the issue was raised in the case of Dolly the sheep, following her death from a contagious lung disease. The claim that Dolly’s early death involved premature senescence has been vigorously contested (e.g. by Kerry Lynn Macintosh in her book, Illegal Beings: Human Clones and the Law), and Dolly’s creator, Dr Ian Wilmut has expressed the view that her illness and death were probably unrelated to the fact that she was a clone. An interesting test would be to make hundreds of clones from an animal near its age of death and then study the resulting lifespans from the offspring clones. Would the offspring clones die an early death? Would there be a higher chance of failure in creating the cloned offspring due to DNA damage over time in the parent?
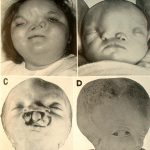
A set of rare hereditary (genetic) disorders, each called progeria, has been known for some time. Sufferers exhibit symptoms resembling accelerated aging, including wrinkled skin. The cause of Hutchinson–Gilford progeria syndrome was reported in the journal Nature in May 2003. This report suggests that DNA damage, not oxidative stress, is the cause of this form of accelerated aging.